
ACD/ADME Suite™ predicts absorption, distribution, metabolism, and excretion (ADME) properties to support drug discovery and development with structure-based calculations.
Vendor
ACD Labs
Company Website
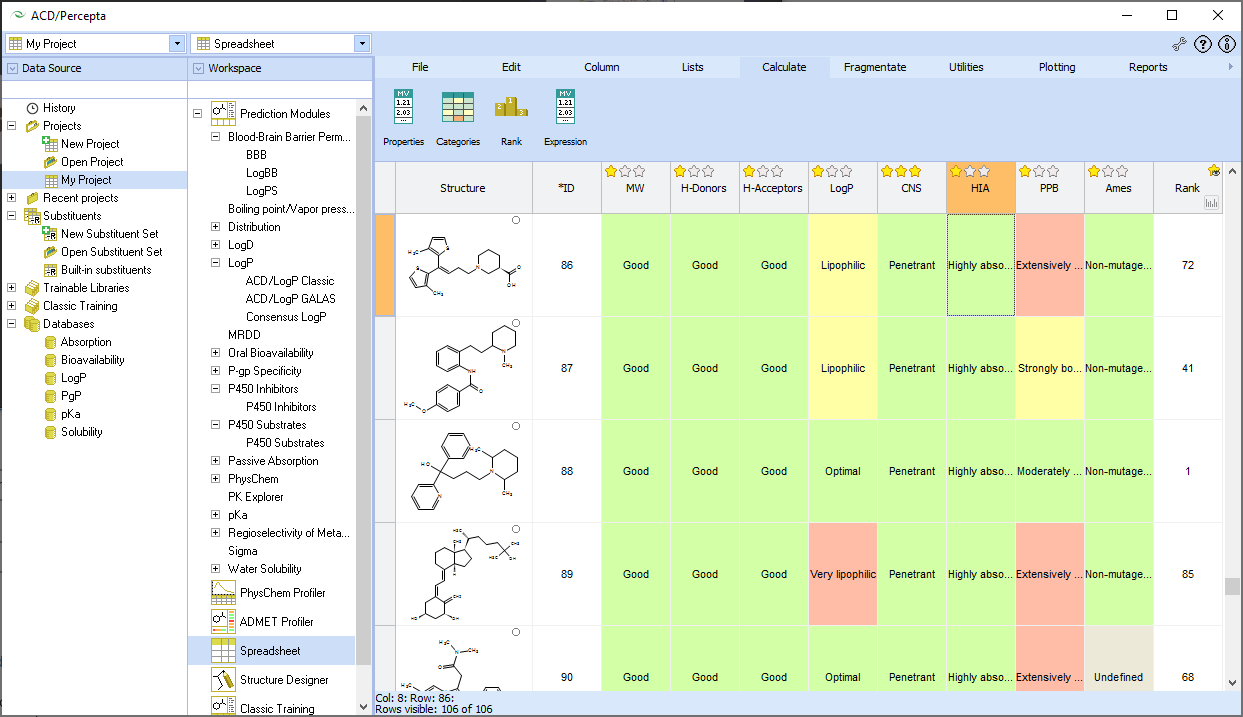
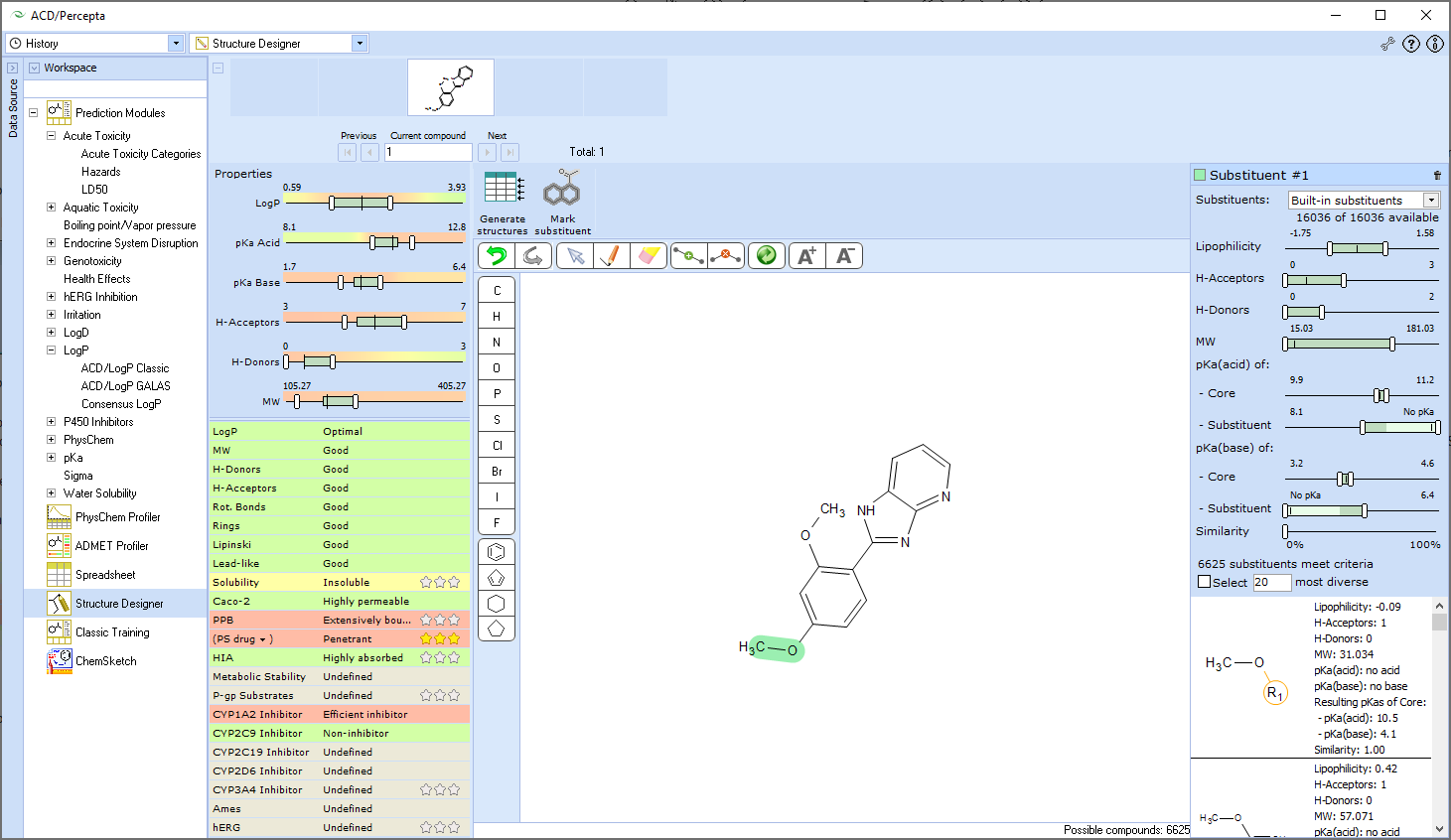
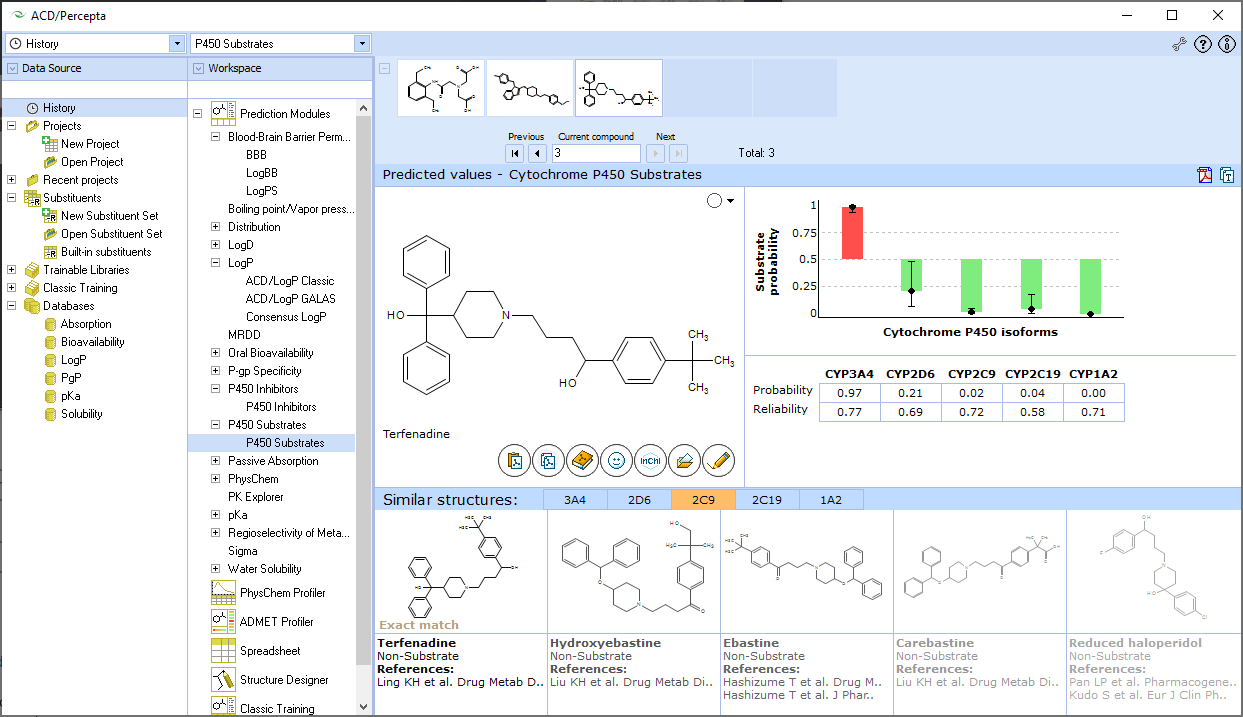
ACD/ADME Suite is a collection of prediction modules that provide structure-based calculations of pharmacokinetic properties. It supports high-throughput screening of compound libraries, provides insights into pharmacological effects, and helps ensure product safety for human use. The suite predicts various ADME properties directly from a compound's structure, including blood-brain barrier penetration, cytochrome P450 inhibition and substrate specificity, distribution, maximum recommended daily dose for human clinical trials, oral bioavailability (and its dependence on logP and bioavailability-dose dependence), passive absorption, P-glycoprotein specificity, physicochemical properties (logP, logD, pKa, aqueous solubility, etc.), and sites of metabolism. The software also assesses the reliability of predicted values, allows users to search an internal library of experimental data for select models, and enables training of prediction models with experimental data to better reflect novel chemical spaces. Custom models and in-house prediction algorithms can also be included.
Features & Benefits
- Easy to Use
- Designed for medicinal, synthetic, and research chemists; no programming or computational chemistry expertise needed.
- Fast, Accurate, Reliable Results
- Quickly calculates properties for single compounds or large libraries, based on curated experimental data, with a reliability index.
- Convenient Visualization
- Visualizes substructure/atomic contributions to property values with color-mapping and user-defined color-coding of results.
- Deeper Insights
- Identifies trends and prioritizes compounds with tools to create scatter plots, browse, filter, sort, and rank libraries.
- Customizable with In-House Data
- Expands the applicability domain of trainable modules using your own experimental data.
- Expandable to Third-Party Models
- Creates a single environment for predicted data by including third-party and in-house models.
- Property-Based Structure Modification and Lead Optimization
- Investigates structure/lead modification to reach a target property profile.
- PK Explorer
- Explore the dependency of various pharmacokinetic parameters on physicochemical properties.
- Visualize the dependence of the following parameters on dose, logP, and pKa (results provided as graphical plots. Use predicted values or enter experimentally derived values for logP and pKa): %F–LogP (the dependence of oral bioavailability on logP at a defined dose) Cp(Max)–LogP (the dependence of maximum achievable drug concentration on logP at a defined dose) %F–Dose (bioavailability–dose relationship) Cp(Max)–Dose (maximum achievable drug plasma levels at different doses) Cp–Time (simulation of plasma concentration-time curves for oral and intravenous administration)
- Estimate absorption (ka), total body clearance (ke), solubility in the gastrointestinal tract (SolGI), volume of distribution (Vd), and presystemic metabolism in the gut and liver (first-pass clearance) based on entered physicochemical property values
- Alter any of these values to recalculate
- Choose to ignore first-pass clearance
- Calculate maximum achievable plasma level (Cp(Max)) and the corresponding time (Tmax), area under the concentration-time curve (AUC) after oral and intravenous administration, and oral bioavailability (%F)
- Explore the dependency of various pharmacokinetic parameters on physicochemical properties.
- New P-gp Efflux Ratio module
- Enables viewing and analyzing quantitative estimates of P-gp efflux potential, including efflux ratio values with contributions of passive diffusion and active P-gp efflux rates
- Significant expansion of the training libraries
- Used in GALAS models of P-gp substrate specificity (81% increase) and Plasma Protein Binding (135% increase) leading to substantial improvements in prediction accuracy
- User friendly heatmap
- Allows you to explore the necessary changes to the molecule’s physicochemical profile to achieve desired transport characteristics
- Significant improvements to pKa, logP, and logD predictions
- Impacting ADME endpoints such as absorption, bioavailability, blood-brain barrier penetration, and pharmacokinetic simulations